China University of Science and Technology uses deep learning to achieve de novo design of protein sequences with high experimental success
2022/7/22
The team of Professor Liu Haiyan and Associate Professor Chen Quan from the School of Life Sciences and Medicine, University of Science and Technology of China, in collaboration with the team of Professor Li Houqiang from the School of Information Science and Technology, developed an algorithm ABACUS-R, which is based on deep learning to design amino acid sequence from the ground up for a given backbone structure. The design success rate and design accuracy of ABACUS-R exceed that of the original statistical energy model ABACUS. The relevant results were published in Nature on July 21, 2022, under the title "Rotamer-Free Protein Sequence Design Based on Deep Learning and Self-Consistency. Computational Science.
The team of Professor Liu Haiyan and Associate Professor Chen Quan is committed to developing data-driven approaches to protein design, and has established and experimentally validated the SCUBA model for designing master chain structure from the ground up using neural network energy functions, as well as the statistical energy function ABACUS for designing amino acid sequence for a given master chain structure. However, the method of sequence design by optimizing the energy function still has some shortcomings in the aspects of success rate and computational efficiency. A number of recent studies have shown that amino acid sequence design using deep learning can exceed the energy function method in calculating indexes such as the recovery rate of natural amino acid residues. However, in the published work, the experimental verification results of the related methods are far from the success rate of the energy function method. The ABACUS-R model reported in this paper not only exceeds ABACUS in calculation index, but also greatly improves the success rate and structural accuracy in experimental verification.
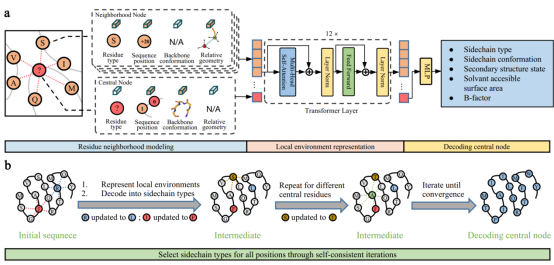
The method for sequence design with ABACUS-R consists of two parts (Figure 1). The first part is a pre-trained encoder-decoder network: The network uses Transformer to map the chemical and spatial structural environment of the central amino acid residue into a hidden space representation vector, and then uses a multi-layer perceptron network to decode the vector into a variety of real features, including the type of the central amino acid residue (Figure 1a). In the second part of the method, after training with non-redundant natural protein sequence structure data, the ABACUS-R encoder-decoder is used to de novo design all or part of the amino acid sequence for a given backbone structure. Specifically, starting from any initial sequence, ABACUS-R encoder-decoder is applied to each type of undetermined residue to obtain the most appropriate residue type dependent on the environment, and the residue types at different sites are iterated to the maximum degree of self-consistency (FIG. 1b).
![]()
Figure 1. Principle of protein sequence design using ABACUS-R model. (a) A pre-trained encoder-decoder network; (b) Complete sequence de novo design using self-consistent iterative strategy.
![]()
Professor Liu Haiyan and Associate Professor Chen Quan from the School of Life Science and Medicine and Professor Li Houqiang from the School of Information Science and Technology are the corresponding authors of this paper. Yufeng Liu, a master student in the School of Life Sciences and Medicine, Zhang Lu, a doctoral student, and Weilun Wang, a doctoral student in the School of Information Science and Technology, are co-first authors of the paper. The research was supported by the Ministry of Science and Technology, the National Natural Science Foundation of China and the Chinese Academy of Sciences.
Source: HKUST News Network

Hefei Zhongke Hanhai Qingzhou Technology Management Co., LTD. 版权所有 Disclaimer 皖ICP备2022016021号-1
Address: Shuangfeng Zhigu Innovation and Entrepreneurship Technology Park, Changfeng County, Hefei City